


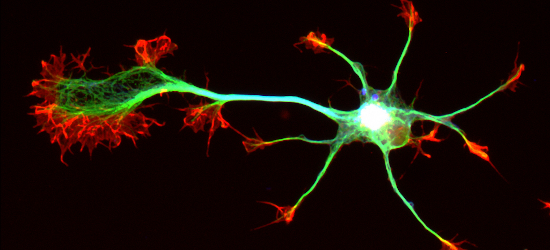
Image microscopique d'un neurone, avec en vert ses prolongements (axones) et en rouge ses synapses
Maladie ne touchant que les filles, le syndrome de Rett a des conséquences neurologiques très graves. L’identification d’une voie moléculaire commune à la maladie de Huntington, autre pathologie génétique rare, apporte des perspectives thérapeutiques encourageantes.
Atteinte inhabituelle et grave du développement cérébral, le syndrome de Rett se caractérise par une forte régression des capacités de l’enfant, après des mois d’évolution normale. Liée à la mutation d’un gène porté par le chromosome sexuel X, cette maladie génétique ne touche que les filles. Pour l’heure, les traitements existants ne soulagent que les symptômes.
C’est pourquoi, développer un traitement spécifique prévenant l’apparition des manifestations de cette maladie rare s’avère crucial. Des chercheurs français de l’Inserm ont réalisé une avancée majeure en ce sens.
Absence de supervitamine neuronale
La protéine codée par le gène muté a une fonction importante dans le développement et le bon fonctionnement des neurones. C’est pourquoi sa mutation entraîne d’importants troubles neurologiques. Comme l’explique un des chercheurs, Jean-Christophe Roux du Centre de génétique médicale de Marseille : « Parmi les voies physiologiques altérées, on trouve notamment celle du facteur BDNF, une sorte de supervitamine ayant un rôle-clé dans la survie, le développement et la plasticité neuronale. »
Cette supervitamine circule habituellement dans les axones (prolongement des neurones), au sein de vésicules. « Dans le syndrome de Rett, poursuit le scientifique, ces vésicules sont rares et lentes, et les neurones sont comme atrophiés et peu connectés. »
Précédemment, dans la maladie de Huntington, l’importance d’une protéine, appelée huntingtine, a été montrée dans le transport de la supervitamine neuronale. Une simple modification de la huntingtine entraîne une accélération de ce transport.
Les chercheurs ont donc utilisé la huntingtine phosphorylée dans un modèle animal reproduisant le syndrome de Rett. Avec succès puisque des souris atteintes ainsi traitées présentent une meilleure fonction respiratoire et locomotrice. Leur poids s’est stabilisé et leur espérance de vie s’est vue prolongée. La huntingtine phosphorylée a donc permis de restaurer en partie la fonctionnalité de la supervitamine.
Il faut maintenant que les scientifiques développent des molécules plus spécifiques et utilisables chez l’enfant atteint du syndrome de Rett. Le chemin est donc encore long, se chiffrant en années, avant les premières études cliniques dans le traitement cette maladie. Mais, pour la première fois, une voix thérapeutique prometteuse a été identifiée.